
Частота заболевания
Причина СМА
СМА – наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN – протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
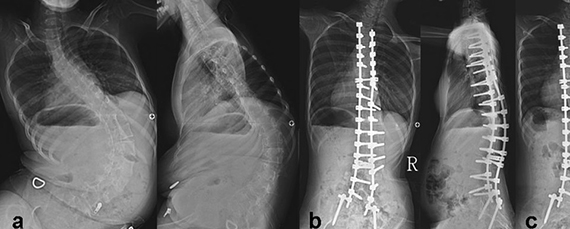
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.
От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена – SMN1 и SMN2.
При этом SMN1 – основной «заказчик» данного белка, а SMN2 – дополнительный, он вырабатывает белок в количестве, недостаточном для нормальной работы организма. В случаях, когда в геноме человека SNM1 отсутствует, SNM2 начинает выполнять замещающие функции, но никогда не может полностью восполнить недостачу.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения – не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III, болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА IV, этот тип называется ещё «взрослой СМА», поскольку болезнь проявляется обычно в возрасте после 35 лет.
Симптомы – мышечная слабость, сколиоз и тремор. Кроме того, развиваются контрактуры суставов (ограничения подвижности в суставах) и нарушения метаболизма.
Прогрессирование заболевания не очень быстрое, сначала мышечная слабость затрагивает мышцы ног, затем – рук. Обычно проблем с глотательной и дыхательной функцией у больных нет.
Большинство из больных IV типом СМА могут ходить, и лишь некоторым приходится прибегать к инвалидным коляскам.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными – они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
В последние годы в мире активно разрабатываются препараты против СМА, три из них уже применяются в мире, остальные находятся на разных стадиях разработки.
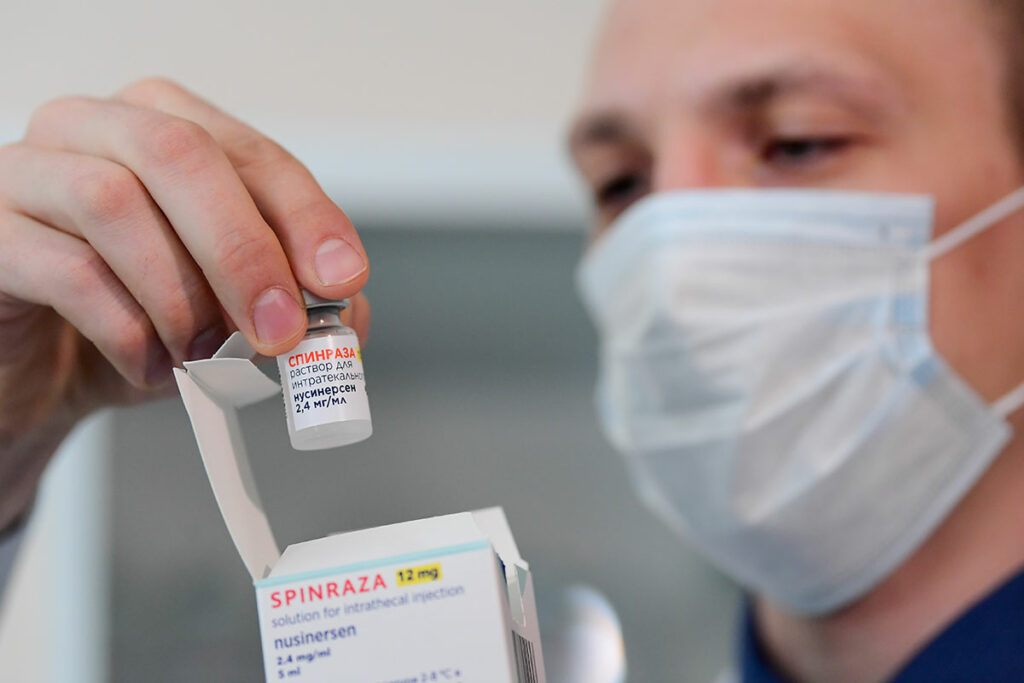
Спинраза – препарат, разработанный компанией Biogen, увеличивает производство белка SMN из «резервного» гена SMN2. При регулярной терапии спинраза приостанавливает развитие СМА и стабилизирует состояние больного.
Спинраза выпускается в дозировке 12 мг на 5 мл для интратекального введения (непосредственно в спинномозговую жидкость) и одобрен для всех возрастов и типов СМА без каких-либо ограничений.
В первый год необходимо будет сделать 6 инъекций: 3 дозы с 14-ти дневным интервалом, 1 дозу спустя 30 дней и далее – раз в 4 месяца. Впоследствии препарат нужно будет применять постоянно в течение всей жизни каждые 4 месяца.
Препарат следует начать применять, как только был поставлен диагноз. Эффективность и безопасность применения нусинерсена изучалась у детей в возрасте от 0 до 17 лет, опыт применения у пациентов старше 18 лет ограничен, применение препарата у пациентов старше 65 лет не изучалось.
После применения спинразы наблюдались улучшения двигательной активности у пациентов со СМА I, II и III типа. Возможные осложнения спинразы – инфекция верхних дыхательных путей, инфекции нижних дыхательных путей и запоры, возможно также развитие ателектаза, отклонения коагуляции и тромбоцитопения, включая острую тяжелую тромбоцитопению, а также развитие гломерулонефрита.
13 февраля 2019 года спинраза получила в России орфанный статус. 16 августа 2019 года препарат был официально зарегистрирован в России.
3 августа 2020 комиссия Минздрава рекомендовала правительству включить препарат для лечения спинальной мышечной атрофии в перечень жизненно необходимых и важнейших лекарственных препаратов на следующий год. Однако по состоянию на сентябрь переговоры по возмещению за счет средств федерального бюджета на 2021 год не завершены.
Есть прецеденты выигранных судов, когда суд обязал региональные власти обеспечить пациентов спинразой за счёт региональных бюджетов.
Стоимость препарата – около 8 миллионов рублей за одну инъекцию.
Золгенсма (Zolgensma) — первый препарат генной терапии, разработанный для лечения спинальной мышечной атрофии (СМА). Предназначен для устранения генетической причины СМА путем замены дефектного или отсутствующего гена SMN1 для остановки прогрессирования заболевания. Препарат доставляет полностью функциональную копию гена SMN в организм человека. Препарат разработан компанией Авексис для однократного применения (одна инъекция на всю жизнь).
В настоящее время препарат одобрен в США для больных СМА возрастом до 2 лет, включая тех, кто не имеет симптомов при постановке диагноза. В Европе препарат получают больные СМА I типа с количеством копий SMN2 не более 3 копий, ограничения по весу 21 кг. Препарат вводится однократно внутривенно, доза определяется с учётом массы тела ребёнка.
Наиболее эффективно применение препарата до появления первых симптомов заболевания у детей, у которых наличие СМА установлено по результатам скрининга. Исследования о применении препарата у детей более позднего возраста, уже имеющих симптомы СМА, продолжаются.
Наиболее частыми нежелательными реакциями на фоне применения Золгенсма были повышение активности печёночных ферментов и рвота. В инструкции по применению препарата имеется предупреждение о риске развития тяжёлого острого поражения печени.
В середине июля 2020 года компания «Новартис» подала в Министерство здравоохранения РФ досье на регистрацию препарата к применению в России. По существующей процедуре, процесс регистрации может занять до полугода.
В настоящее время компанией-производителем объявлена программа сострадательного применения (до официальной регистрации препарата), по которому 100 доз препарата будут распространены между ста пациентами со СМА любого типа до двух лет методом лотереи. В России по этой программе препарат получили 4 ребёнка.
Кроме того, за счёт средств благотворителей препарат получили 16 российских детей, ещё двое принимают участие в программе клинических испытаний.
Стоимость дозы препарата составляет около 152 миллионов рублей (самый дорогой препарат в мире).
Рисдиплам (RG7916) («Эврисди») – препарат компании Roche, является модификатором сплайсинга (генетической модификации) гена SMN2, увеличивающим экспрессию полноразмерных функциональных белков. В отличие от других препаратов, применяется перорально.
Исследования проводились с участием пациентов от 0 до 60 лет со СМА II и III типов. Улучшает моторную функцию пациентов. Противопоказаний на сегодняшний день не зарегистрировано.
18 марта 2020 года компания «Рош» подала заявку на рассмотрение для дальнейшей регистрации препарата в Министерство здравоохранения РФ.
В настоящее время производителем открыта программа дорегистрационного доступа к препарату.
Кроме того, в мире идут клинические испытания ещё нескольких препаратов – бранаплам, релдесемтив, SRK-015 и других.
По мнению врачей комбинация препаратов при лечении СМА возможна, но её целесообразность должна рассматриваться в каждом случае индивидуально.
Можно ли помочь больным СМА и как именно?

Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Понять, есть ли связь между СМА и прививками, может объяснение разницы между СМА и полиомиелитом. Полиомиелит – инфекционное заболевание, когда от инфекции повреждается организм изначально здорового ребёнка. Ребёнок со СМА, родившийся с повреждённым геномом, внешне может выглядеть здоровым, но на самом деле он уже болен, просто симптомы его болезни проявляются постепенно. В этом отношении СМА – такая же «отложенная» болезнь как, например, миодистрофия Дюшенна или синдром Ретта, когда ребёнок, некоторое время развивавшийся в соответствии с нормой, теряет приобретённые ранее навыки и становится инвалидом.
Большинство проявлений СМА связаны с освоением первых двигательных навыков. Первые проявления болезни совпадают по времени с несколькими возрастными прививками. В итоге человек и его родные могут утверждать, что он «заболел от прививки», но на самом деле у него просто проявились признаки болезни, которая уже была.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Отсутствие специальной диагностики привело также к путанице в диагнозах. Большинство больных СМА в России не выявлены, у многих выявленных в качестве диагноза записана «болезнь Верднига-Гоффмана», хотя не у всех из них (особенно взрослых) в действительности именно этот тип болезни.
Сколько больных СМА в России?
С учётом частоты заболевания, количество больных СМА в России должно составлять от семи до двадцати четырёх тысяч человек. На сегодняшний день в реестре пациентов фонда «Семьи СМА» находится около 400 человек.
Кто в России помогает людям со СМА и их семьям
Благотворительный фонд «Вера», детский хоспис «Дом с маяком», благотворительный фонд «Детский паллиатив», благотворительный фонд «Семьи СМА», детская паллиативная служба «Милосердие».
С 2014 года в Москве развивается совместный проект службы «Милосердие» и фонда «Семьи СМА» «Клиники СМА» На встречах, которые проходят раз в месяц, больные могут получить консультации пульмонолога, ортопеда, физиотерапевта и психолога. В последнее время часть встреч ориентированы и на нужды взрослых пациентов.
Известные люди со СМА

Итальянка Симона Спиноглио родилась с наследственным заболеванием – спинальной мышечной атрофией 2 типа. Она с самого рождения не может ходить и передвигается только с помощью электрической коляски. Но ее жизнь полна и насыщенна; ничто не может помешать ее стремлению жить.
Симона работает на «горячей линии» итальянской Ассоциации «Семьи СМА» (Famiglies of SMA) и помогает детям и взрослым со СМА и другими нервно-мышечными заболеваниями.
Также Симона записала несколько популярных в итальянском сообществе СМА песен – о свободе делать то, что ты хочешь, несмотря на болезнь.
Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
В 2008 году собрала собственную музыкальную группу (распалась в 2010). В 2013 году приняла участие в конкурсе «Фактор А» на телеканале «Россия». Заняла второе место и получила персональную премию Аллы Пугачёвой «Золотая звезда Аллы». В 2017 году из-за недопуска России в конкурсную программу не смогла принять участие в конкурсе «Евровидение». Передвигается на коляске.
Программист из Владимира Валерий Спиридонов. Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).
Сегодня Валерий – член городской общественной палаты Владимира, эксперт по вопросам доступной среды, а также создатель собственного сообщества «Desire for life», рассказывающего о создании доступной среды и перспективных медицинских проектах. Валерий – участник многих телепрограмм на российском и зарубежном ТВ.
Полезные ресурсы про СМА
Американская ассоциация мышечных дистрофий (англоязычный),
Форум о миопатиях (российский).
Сайт о миопатиях (Беларусь):
Фонд «Дети со СМА» (Украина) (на сайте есть форум).
Благодарим за предоставленную информацию фонд «Семьи СМА» и лично Ирину Старову-Кислину.
Мы просим подписаться на небольшой, но регулярный платеж в пользу нашего сайта. Милосердие.ru работает благодаря добровольным пожертвованиям наших читателей. На командировки, съемки, зарплаты редакторов, журналистов и техническую поддержку сайта нужны средства.
